



DeepSomatic employs advanced AI to precisely identify cancer-driving mutations within a tumor's genetic sequence, significantly enhancing diagnostic precision and treatment strategy development.
Cancer, fundamentally a genetic disease, arises from dysregulated cellular division controls. Its diverse forms present unique challenges due to distinct genetic underpinnings. Accurately identifying genetic mutations within tumor cells is paramount for understanding cancer and formulating targeted treatment plans. Modern clinical practice increasingly relies on sequencing tumor cell genomes to guide therapies that specifically inhibit cancer progression.
In collaboration with UC Santa Cruz Genomics Institute and leading federal and academic researchers, our breakthrough paper, “DeepSomatic: Accurate somatic small variant discovery for multiple sequencing technologies”, published in Nature Biotechnology, introduces a revolutionary AI-powered tool. This system leverages convolutional neural networks to achieve superior accuracy in identifying tumor variants compared to existing methodologies. DeepSomatic demonstrates remarkable flexibility, effectively processing data from all major sequencing platforms, accommodating diverse sample preparation techniques, and extending its predictive capabilities to novel cancer types beyond its initial training.
We are committed to accelerating cancer research and advancing precision medicine. To that end, we have made both the DeepSomatic tool and our high-quality training dataset publicly available. This initiative complements Google's extensive efforts in developing AI for cancer understanding and treatment, including AI for mammogram analysis in breast cancer screening, CT scan analysis for lung cancer screening, and AI partnerships to advance gynecological cancer research.
Genetic variation acquired after birth
Genome sequencing is indispensable in research and clinical settings for identifying genetic variations against the established human reference genome. Differentiating genuine variants from sequencing artifacts poses a significant challenge. Recognizing this, Google Research pioneered DeepVariant a decade ago, a highly accurate deep neural network for identifying inherited variants, also known as germline variants, present in all cells and inherited from parents.
The genetic landscape of cancer is considerably more intricate, frequently driven by variants acquired post-birth. Environmental factors like UV radiation and chemical carcinogens, alongside spontaneous DNA replication errors, can induce new variants in somatic cells. These acquired mutations can disrupt normal cellular behavior, leading to uncontrolled replication and initiating cancer development, and subsequently driving its progression into more aggressive stages.
Detecting variants unique to specific somatic cell populations is substantially more challenging than identifying inherited variations. Tumor cells exhibit a broad spectrum of acquired variants at varying frequencies, and sequencing error rates can exceed the prevalence of somatic variants within a sample.
Training DeepSomatic to spot genetic variation in tumor cells
We engineered DeepSomatic to overcome these complexities and deliver precise somatic variant identification. In typical clinical and research scenarios, cancer is analyzed by sequencing both tumor cells obtained via biopsy and unaffected normal cells containing germline variations. DeepSomatic is meticulously trained to distinguish tumor-specific variations from inherited ones, providing crucial insights into the mutations driving tumor growth. Furthermore, DeepSomatic excels in tumor-only analysis, enabling variant detection even when a normal cell sequence is unavailable, a critical capability for hematological malignancies like leukemia where isolating purely normal cells from blood samples is difficult. This adaptability across diverse use cases, reflecting common clinical and research methodologies, ensures DeepSomatic's broad applicability.
Mirroring the approach of DeepVariant, DeepSomatic transforms genetic sequencing data into a series of interpretable images. These images encapsulate sequencing data, chromosomal alignment, quality metrics, and other vital variables. DeepSomatic then employs its sophisticated convolutional neural network, processing data from both tumor and non-cancerous cells, to accurately differentiate between the reference genome, an individual's germline variants, and cancer-driven somatic variants, while effectively filtering out sequencing-induced errors. The output is a precise list of cancer-related variants, or mutations.
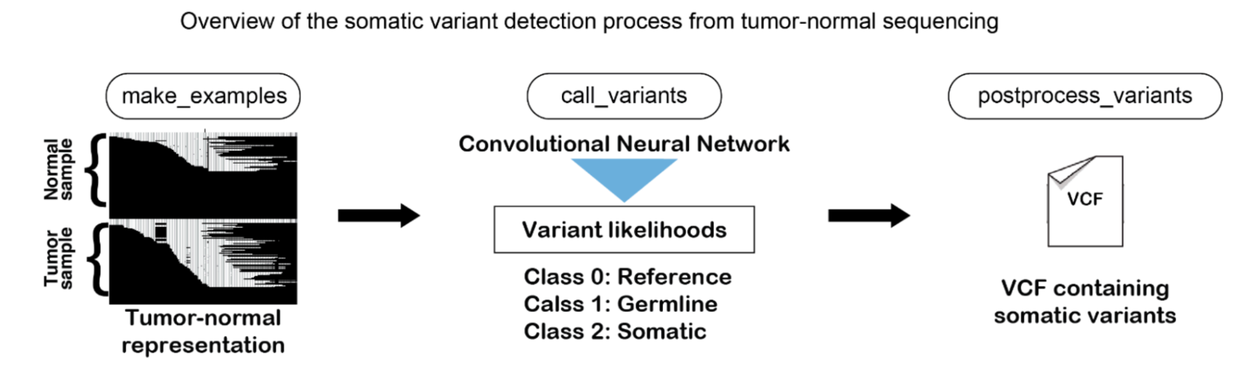
DeepSomatic precisely detects cancer variants in genomic data. The process involves converting tumor and non-cancerous cell sequencing data into images, which are then analyzed by its convolutional neural network. This allows DeepSomatic to distinguish the reference genome, germline variants, and cancer-causing somatic variants, while filtering out sequencing errors, yielding an accurate list of mutations.
Developing accurate models for genetic variation detection across diverse cancer types necessitates comprehensive, high-fidelity data and truth sets. To this end, we have established a novel training and evaluation dataset for tumor variant detection. In partnership with UC Santa Cruz and the National Cancer Institute, we performed whole-genome sequencing on six samples from breast and lung cancer cell lines, analyzing both tumor and adjacent normal cells.
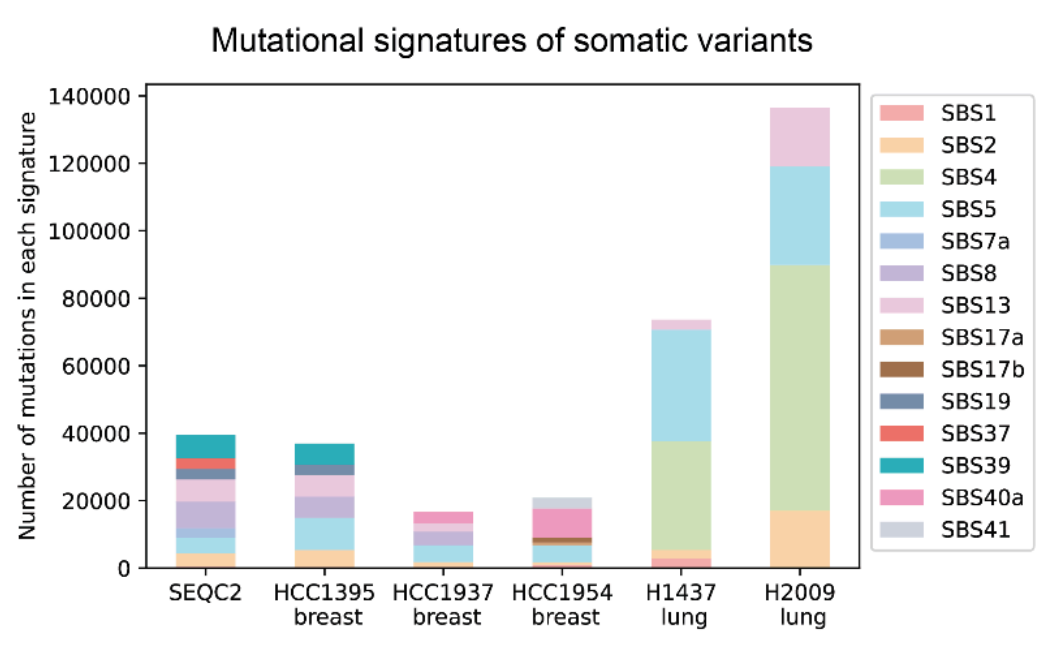
This benchmark dataset, comprising four breast and two lung cancer samples, trains DeepSomatic. Each bar represents mutation counts, with color indicating mutation types. Notably, lung cancer exhibits a distinctive mutation signature (green, SBS4) linked to environmental toxins. Significant inter-individual variation in mutational signatures, even within the same cancer type, can predict treatment response.
To construct a highly accurate training dataset, we employed three leading sequencing platforms: Illumina's short-read sequencing, PacBio's long-read sequencing, and Oxford Nanopore Technology's long-read sequencing. By integrating output from all three, we mitigated platform-specific errors, establishing a unified, precise reference dataset known as the Cancer Standards Long-read Evaluation dataset (CASTLE), for assessing genetic diversity in tumor and normal cells.
Testing DeepSomatic’s ability to spot cancer-related variants
We trained DeepSomatic using three breast cancer genomes and two lung cancer genomes from the CASTLE reference dataset. Subsequent testing involved evaluating its performance on a held-out breast cancer genome and on chromosome 1 from each sample, which were excluded from training.
Our findings reveal that DeepSomatic models, specifically developed for each of the three major sequencing platforms, significantly outperform existing methods in identifying tumor variants with enhanced accuracy. For short-read sequencing, we benchmarked against SomaticSniper, MuTect2, and Strelka2, with SomaticSniper used for single nucleotide variants (SNVs). For long-read sequencing, comparisons were made with ClairS, a deep learning model trained on synthetic data.
Across the six reference cell lines and a seventh preserved sample, DeepSomatic identified 329,011 somatic variants. It demonstrated particular proficiency in detecting cancer variations involving insertions and deletions (Indels), substantially improving the F1-score—a metric balancing recall and precision. For Indels on Illumina data, DeepSomatic achieved 90% accuracy compared to the next best method's 80%. On Pacific Biosciences sequencing data, DeepSomatic's accuracy exceeded 80%, a dramatic improvement over the next best method's less than 50%.

DeepSomatic's performance (purple) on a standard breast cancer research sample, compared to alternative tools. While multiple software options exist for Illumina data, only one (pink) effectively handles long-read data from PacBio and Oxford Nanopore. DeepSomatic shows marginal improvement for single nucleotide variants and significant gains for Indels.
The seventh sample, a preserved breast cancer tumor sample treated with formalin-fixed-paraffin-embedded (FFPE)—a common preservation method that can introduce DNA damage complexities—was also analyzed using whole exome sequencing (WES), a cost-effective approach focusing on protein-coding regions. When DeepSomatic was trained on FFPE and WES data, and subsequently tested on chromosome 1 (excluded from training), it surpassed other tools. This indicates its potential for analyzing lower-quality or historical tumor samples, even those sequenced only via exome, and its applicability to clinical datasets.

DeepSomatic exhibits significantly higher accuracy on challenging samples: formalin-fixed paraffin embedded (FFPE) tissues (left), a standard preservation method, and whole exome sequencing (WES), which targets protein-coding regions (right). The central section illustrates a sample processed with both FFPE and WES.
Applying DeepSomatic to other cancers
To evaluate DeepSomatic's performance on diverse cancer types, we analyzed a glioblastoma sample, an aggressive brain cancer driven by a small number of key mutations. DeepSomatic successfully identified these variants, demonstrating its capability to generalize its learning to distinct cancer types.
In collaboration with Children's Mercy in Kansas City, we also analyzed eight previously sequenced pediatric leukemia samples. As leukemia affects the bloodstream, obtaining a purely normal blood sample is unfeasible. Despite this constraint, DeepSomatic accurately identified known variants and discovered 10 novel ones, proving its efficacy in tumor-only analysis scenarios.
What’s next
Our vision is for research laboratories and clinicians to widely adopt DeepSomatic. Detecting established cancer variants can guide treatment choices, including chemotherapy, immunotherapy, and other modalities. The identification of novel mutations holds the potential to unlock entirely new therapeutic avenues. We aim to empower researchers and clinicians to gain deeper insights into individual tumors, elucidate their underlying drivers, and ultimately deliver the most effective treatments to patients.
Acknowledgments
We extend our sincere gratitude to all research participants whose contributions and cell line donations were instrumental to this work and broader biomedical research. We acknowledge our collaborators at UC Santa Cruz Genomics Institute, the National Cancer Institute, the Frederick National Laboratory for Cancer Research, Children’s Mercy Hospital, and NYU. Special thanks to Hannah Hickey for writing contributions. We also thank Avinatan Hassidim, Katherine Chou, Lizzie Dorfman, and Yossi Matias for their invaluable research leadership support, and Resham Parikh and Isha Mishra for their communications support.